Annotating scFv Libraries
Single-chain variable fragments (scFv) are engineered proteins that fuse the variable regions of heavy (VH) and light (VL) antibody chains into a single polypeptide, connected by a flexible peptide linker. Analyzing scFv sequencing data is crucial for antibody discovery and engineering.
This guide will walk you through the process of annotating scFv NGS data using the MiXCR scFv Alignment block on Platforma. This powerful tool can handle various data types, including:
- Long-read data (e.g., PacBio, Oxford Nanopore) that covers the full scFv construct.
- Short-read paired-end data (e.g., Illumina) that covers partial fragments of the construct.
- Data with Unique Molecular Identifiers (UMIs) for read deduplication and error correction.
After alignment, we will explore how to browse, filter, and analyze the resulting clonotypes using the Clonotype Browser block.
Project setup
Before beginning the analysis, ensure your project is properly set up and you have added your raw sequencing datasets. See the Data Import Guide for details.
Getting started: MiXCR scFv alignment
The entire workflow is managed by the MiXCR scFv Alignment block.
- From your project's
Samples & Datapage, click Add Block. - Use the search bar to find and select MiXCR scFv Alignment.
- Click Add To Project to add the analysis block to your workspace.
Settings configuration
Correctly configuring the settings is the most critical part of the analysis. Below is a breakdown of the key parameters.
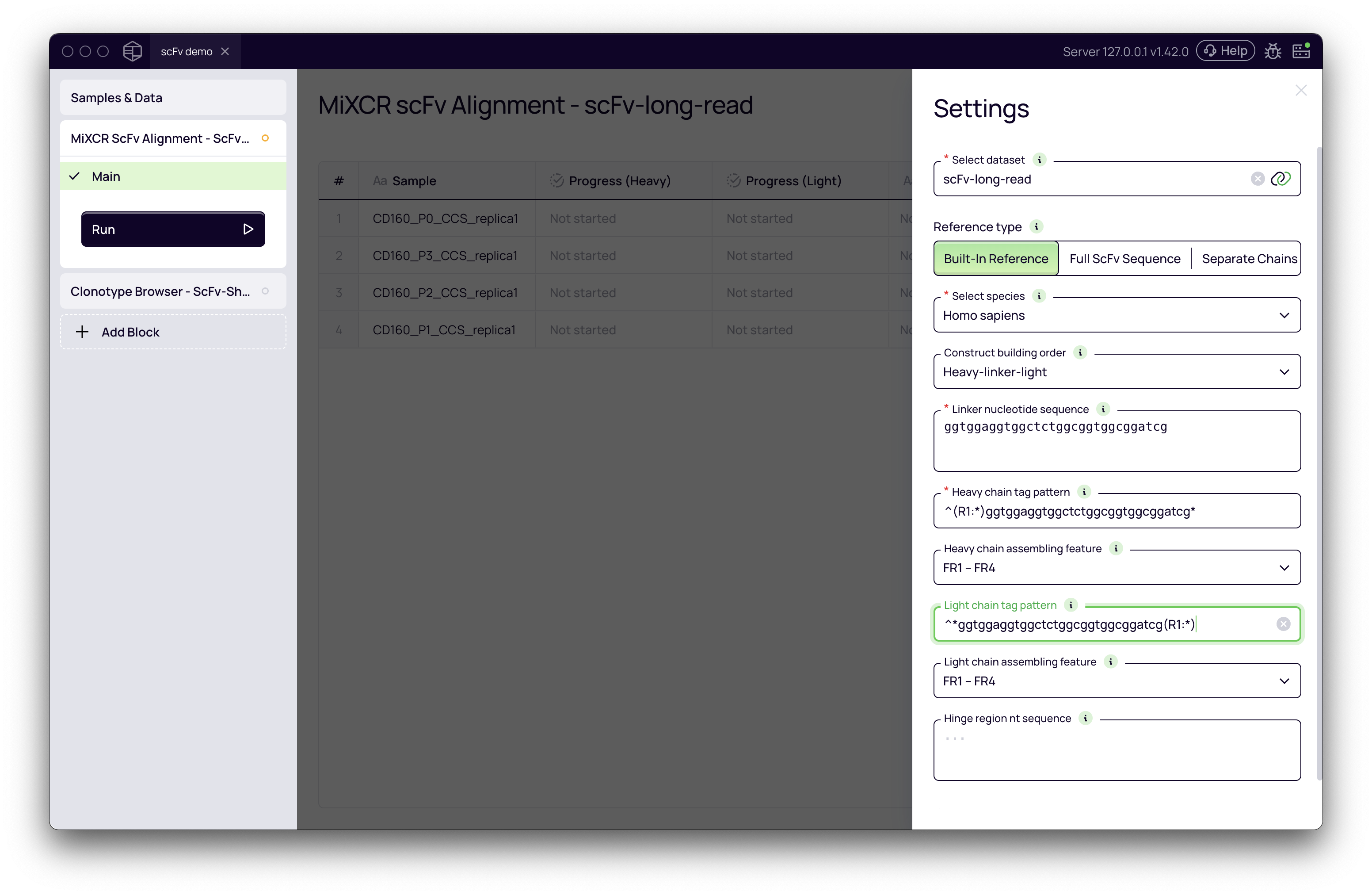
Linker nucleotide sequence
The linker is a short peptide sequence that connects the VH and VL domains. Its nucleotide sequence is a crucial piece of information that you must provide, as MiXCR uses it to locate the heavy and light chains within your sequencing reads.
A common linker is the (G₄S) repeated few times, for example ggtggaggtggctctggcggtggcggatcg. However, many variants exist, so it is essential to use the exact sequence for the linker in your specific scFv library.
Tag patterns
Tag patterns are rules that tell MiXCR how to parse your sequencing reads. They define where the heavy chain, light chain, and UMIs (if any) are located relative to the linker.
The syntax for these patterns can be complex. For a comprehensive guide, refer to the official MiXCR documentation.
Below, we provide tested examples for common scenarios.
Examples for common library types
Here are complete parameter sets for three common scFv library types.
1. Long-read data (e.g., PacBio)
This example is for full-length scFv reads where the entire construct is sequenced in Read 1 (R1).
- Construct building order:
Heavy-linker-light - Linker nucleotide sequence:
ggtggaggtggctctggcggtggcggatcg - Heavy chain tag pattern:
^(R1:*)ggtggaggtggctctggcggtggcggatcg*- This pattern captures the heavy chain from the start of Read 1
^(R1:*)up to the linker sequence.
- This pattern captures the heavy chain from the start of Read 1
- Light chain tag pattern:
^*ggtggaggtggctctggcggtggcggatcg(R1:*)- This pattern finds the linker and captures the sequence that follows it as the light chain
(R1:*).
- This pattern finds the linker and captures the sequence that follows it as the light chain
- Heavy chain assembling feature:
FR1 - FR4 - Light chain assembling feature:
FR1 - FR4
2. Short-read paired-end data (e.g., Illumina)
This is for short-read data where the heavy chain is in Read 1 and the light chain is in Read 2.
- Construct building order:
Heavy-linker-light - Linker nucleotide sequence:
ggaggcggcggttcaggcggaggtggctctggcggtggcggaagt - Heavy chain tag pattern:
^(R1:*)ggaggcggcggttcaggcggaggtggctctggcggtggcggaagt*\^*- This extracts the heavy chain from the beginning of R1. The
\^*part tells MiXCR that R2 exists but should be ignored for the heavy chain analysis.
- This extracts the heavy chain from the beginning of R1. The
- Light chain tag pattern:
^*ggaggcggcggttcaggcggaggtggctctggcggtggcggaagt(R1:*)\^(R2:*)- This pattern assembles the light chain by combining the end of R1 (after the linker) with the entirety of R2.
- Heavy chain assembling feature:
CDR3 - FR4 - Light chain assembling feature:
FR1 - FR4
3. Short-read data with UMIs
This is similar to the previous example but includes Unique Molecular Identifiers (UMIs) for more accurate quantification.
- Construct building order:
Heavy-linker-light - Linker nucleotide sequence:
ggaggcggcggttcaggcggaggtggctctggcggtggcggaagt - Heavy chain tag pattern:
^(UMI:N{12})(R1:*)ggaggcggcggttcaggcggaggtggctctggcggtggcggaagt*\^*- The
(UMI:N{12})tag at the beginning specifies that the first 12 nucleotides are the UMI. Adjust the number12to match your UMI length.
- The
- Light chain tag pattern:
^(UMI:N{12})*ggaggcggcggttcaggcggaggtggctctggcggtggcggaagt(R1:*)\^(R2:*)- The same UMI tag must be included in the light chain pattern for proper pairing.
- Heavy chain assembling feature:
CDR3 - FR4 - Light chain assembling feature:
FR1 - FR4
Running and interpreting the results
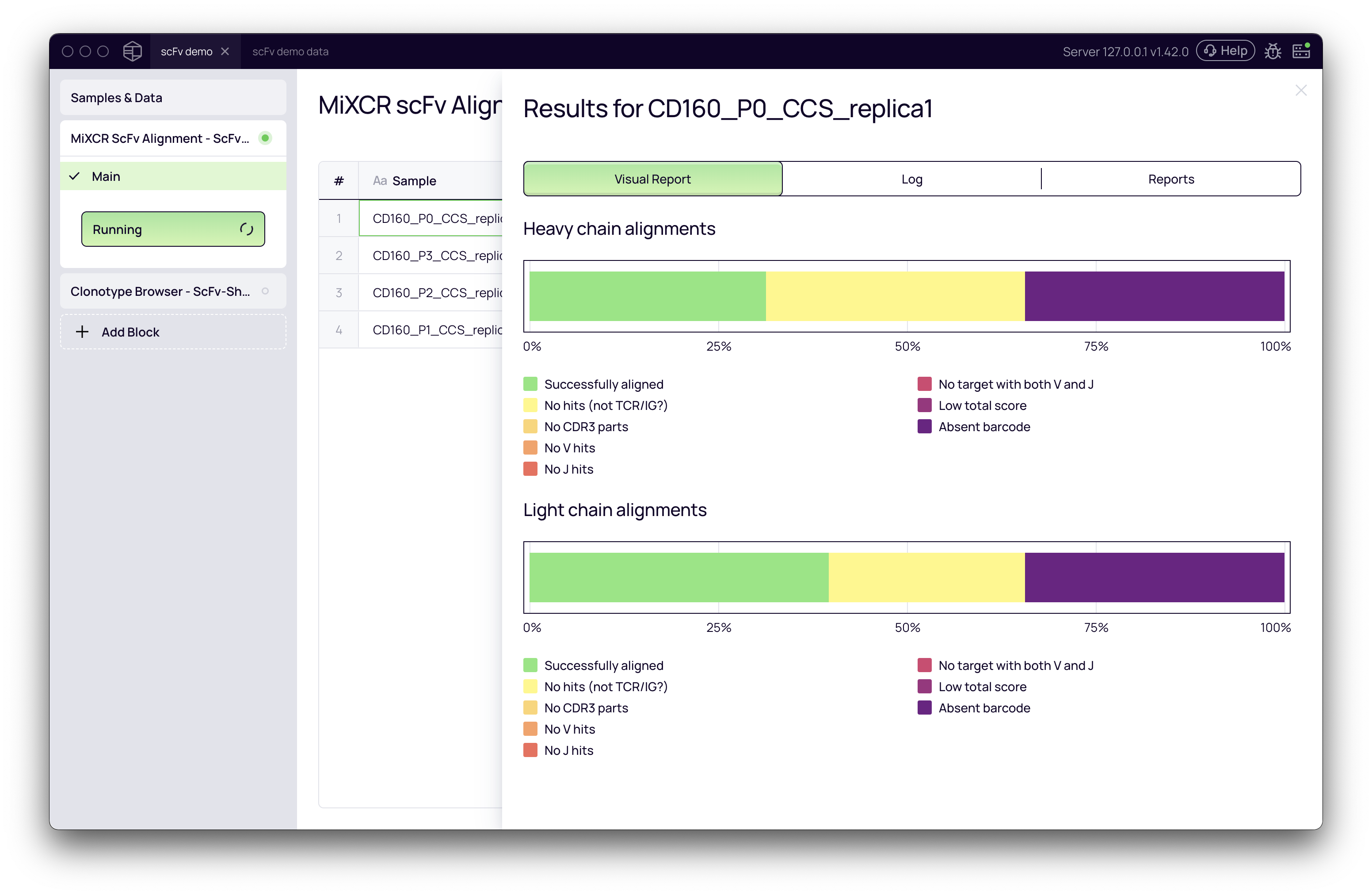
Once configured, click Run. After the analysis status changes to "Done," you can inspect the results by clicking on a sample. The Visual Report tab provides an overview of alignment quality, including the percentage of reads that were successfully aligned, had an absent barcode (linker not found), or produced no hits against the reference database.
Exploring results with clonotype browser
The Clonotype Browser block allows you to dive deep into the aligned and annotated scFv sequences.
- Add a Clonotype Browser block to your project.
- In its Settings, select the output from your completed
MiXCR scFv Alignmentblock. - Click Run to load the data.
Navigating the browser
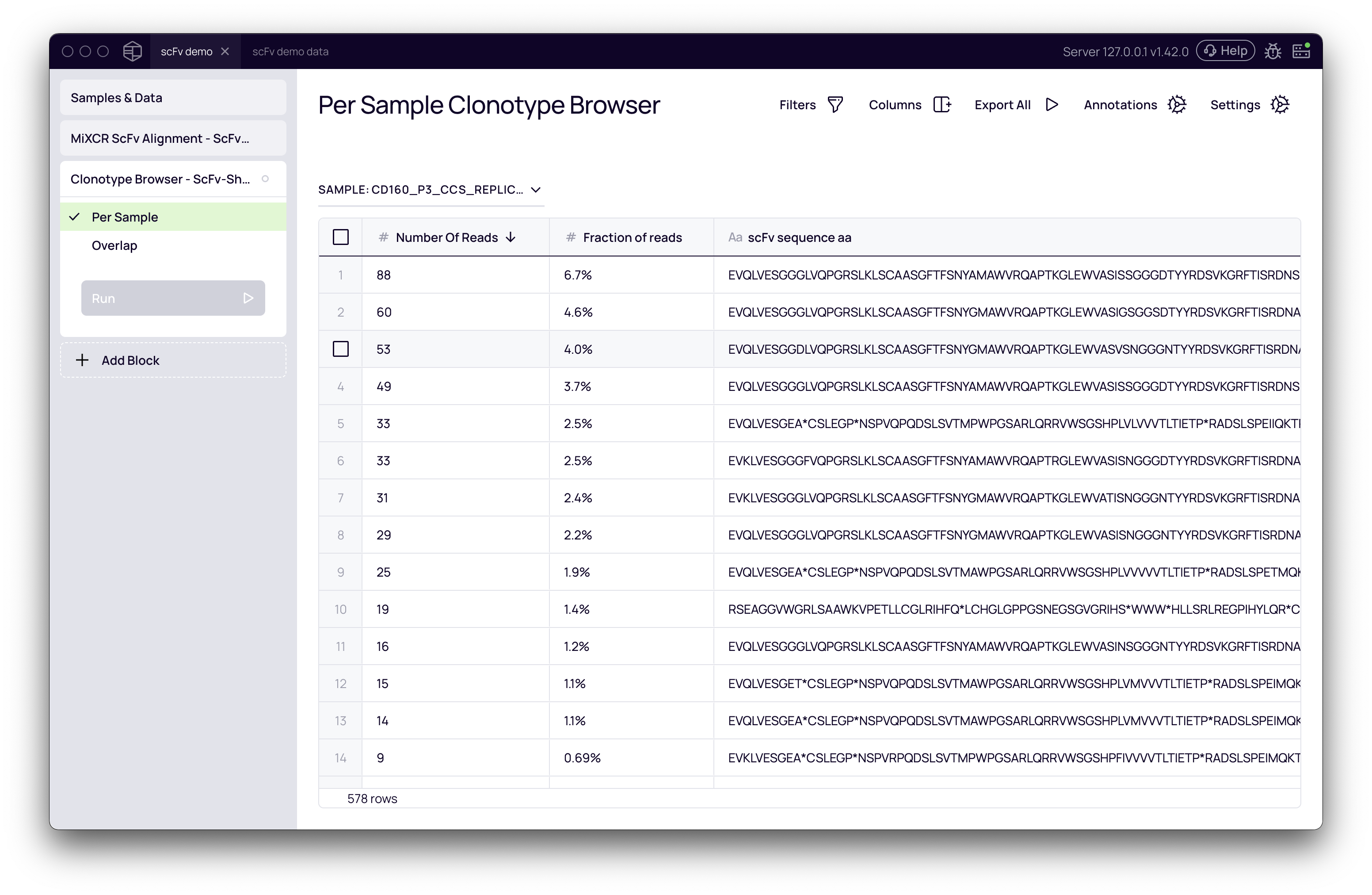
The browser displays a comprehensive table of all identified clonotypes. Key columns include:
- Number of Reads / Number of UMIs: The abundance of each clonotype.
- scFv sequence aa: The full-length, translated amino acid sequence of the scFv.
- Note: For short-read data, where sequencing doesn't cover the full construct, MiXCR imputes the missing regions based on the best-matching germline gene. This reconstruction is a key feature of the tool.
- Heavy/Light Best V/D/J gene: The top germline gene matches for each chain.
You can use the Columns manager to show additional information, such as the nucleotide sequence (scFv sequence nt). In the nucleotide view, imputed bases are shown in lowercase, while directly sequenced bases are in uppercase.
Use the Filters to narrow down your results, for instance, by filtering for productive, in-frame sequences where Productive is True.