Antibody Sequence Liabilities Assessment
In antibody discovery, identifying promising candidates goes beyond just their binding affinity. It's crucial to also assess their potential for manufacturability, stability, and safety. The developability of an antibody is a critical factor that determines its likelihood of becoming a successful therapeutic. This refers to a set of physicochemical properties that make an antibody amenable to being manufactured at a large scale, formulated for stability, and safe for administration to patients.
A key aspect of developability is the assessment of sequence liabilities — specific amino acid motifs within the antibody sequence that can predispose it to undesirable chemical or physical modifications. These modifications can lead to a range of issues, including:
- Loss of potency: Modifications in the antigen-binding regions (CDRs) can reduce or eliminate the antibody's ability to bind to its target.
- Reduced stability: Liabilities can compromise the structural integrity of the antibody, leading to aggregation, fragmentation, and a shorter shelf-life.
- Safety concerns: Modified or aggregated antibodies can trigger an immune response in patients, a phenomenon known as immunogenicity.
The Antibody Sequence Liabilities block in Platforma.bio screens each antibody sequence for known liability motifs and classifies them by fixability — how tractable each liability is to engineer away. This lets you distinguish candidates that need a quick conservative substitution from those that require significant reengineering or are simply not viable.
Prerequisites
Before starting, ensure you have set up a project and samples from each round of your selection experiment have been imported and processed with a clonotyping block.
How to assess antibody sequence liabilities
This analysis is typically performed after you have processed your raw sequencing data, performed clonotyping, and identified enriched clusters.
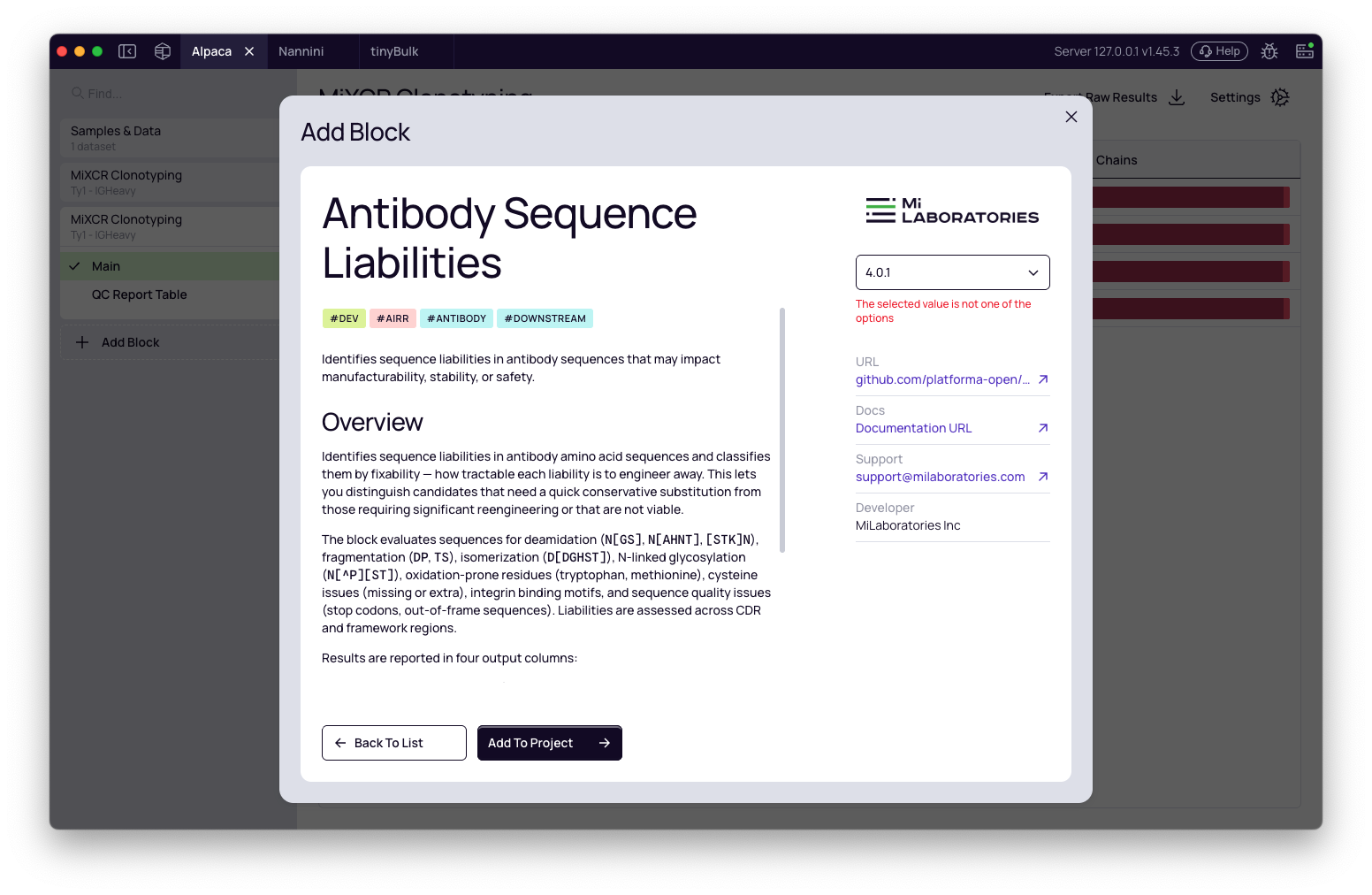
Add the analysis block
Start from your project's analysis pipeline.
- Click the + Add Block button on the left-hand panel.
- In the "Add Block" window, find and select the Antibody Sequence Liabilities block.
- Click Add to Project to add it to your pipeline.
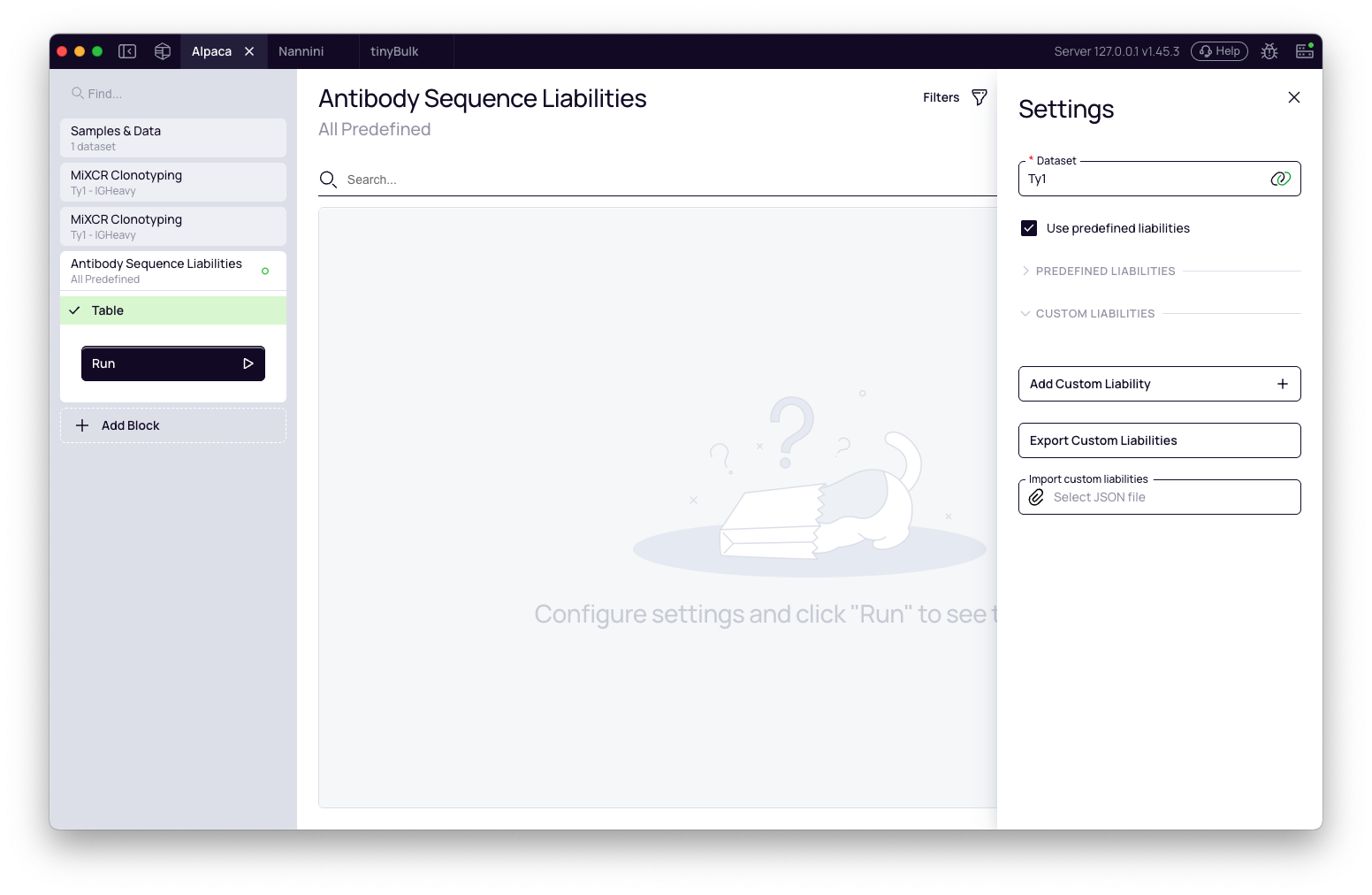
Configure the settings
Once the block is added, configure its settings before running the analysis.
-
Select dataset: Use the dropdown to choose the input dataset — the output of a preceding block that contains your list of clonotypes, such as Redefine Clonotypes.
-
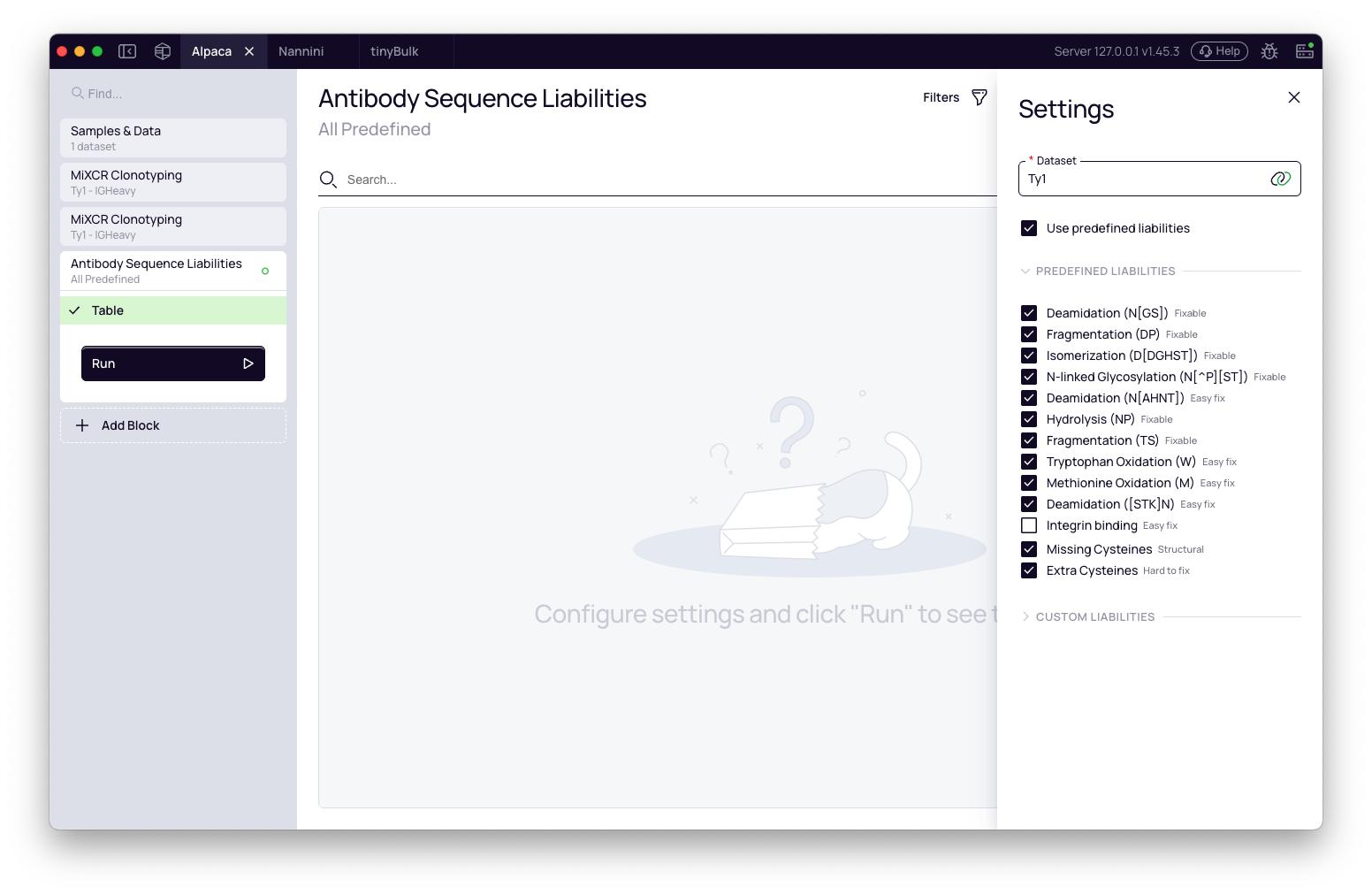
Predefined liabilities: A collapsible section lists all built-in liability definitions. Expand it to inspect each entry — its pattern, risk level, and fixability class. All predefined liabilities are active by default with the exception of Integrin binding (relevant primarily for therapeutic candidates going in vivo). Uncheck any liabilities you want to exclude.
To run only your custom liabilities and skip the predefined set entirely, uncheck Use predefined liabilities.
-
Custom liabilities: Add your own motifs using the Add button. Each custom liability requires a name, a regex pattern, a risk level, a fixability class, and the regions it should be applied to. See Custom liabilities below for details.
-
Click Run to start the analysis.
Interpreting the results
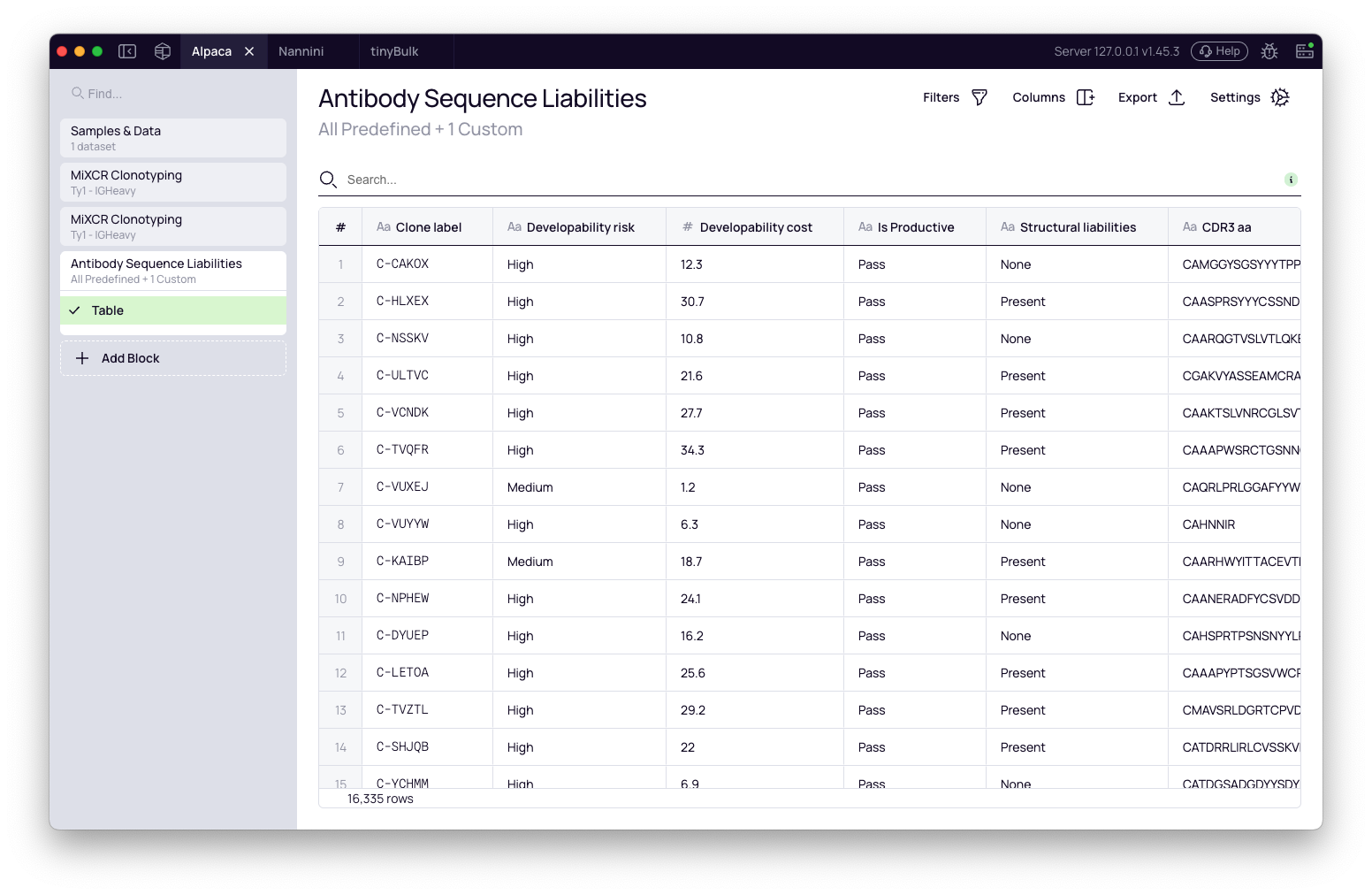
After the block finishes, it generates a table with one row per clonotype and several liability columns.
Score columns
Four columns replace the old single "Liabilities risk" column, each capturing a distinct aspect of the liability picture:
| Column | Values | What it means |
|---|---|---|
| Is Productive | Pass / Fail | Fails when a stop codon or out-of-frame sequence is found. These are not engineering problems — the sequence is invalid. Hard exclusion in Lead Selection. |
| Structural liabilities | None / Present | Present when a structurally disqualifying liability is found — missing conserved cysteines or extra unpaired cysteines. Cannot be fixed by simple substitution. Hard exclusion in Lead Selection. |
| Developability risk | None / Low / Medium / High | The severity of fixable liabilities only (PTM motifs, fragmentation sites). Independent of structural liabilities. Default Lead Selection filter passes None/Low/Medium; High is filtered by default. |
| Developability cost | continuous float | Sum of engineering effort across all non-disqualifying liability hits, weighted by fixability class and region importance. Lower = easier to engineer. Used for ranking, not filtering. |
The columns are independent: a sequence can have Structural liabilities: Present and Developability risk: High simultaneously if it carries both a missing cysteine and a deamidation motif. Each column surfaces its concern separately.
Per-region columns
The block also outputs per-region liability strings (CDR1 aa liabilities, CDR2 aa liabilities, etc.) and per-region risk levels (CDR1 aa risk, etc.). These show which specific motifs were detected in each region, useful for understanding exactly where a liability sits and how risky it is to engineer around.
Per-region risk reflects fixable liabilities only — structural and disqualifying issues are captured in the global score columns, not the regional risk.
Developability cost weights
The developability cost weights each liability hit by two factors:
Fixability weight — how hard the liability is to fix:
| Class | Weight |
|---|---|
| Easily fixable | 1 |
| Fixable | 3 |
| Hard to fix | 8 |
| Structural | 20 |
Region weight — how much the region contributes to binding:
| Region | Weight |
|---|---|
| CDR3 | 1.5 |
| CDR1, CDR2 | 1.2 |
| FR1 | 1.0 |
| FR2, FR3 | 0.5 |
| FR4 | 0.3 |
A deamidation motif (fixability weight 3) in CDR3 (region weight 1.5) contributes 4.5 to the score. The same motif in FR4 contributes 0.9. This reflects the real engineering risk: fixing a CDR3 liability requires more care and has a higher chance of affinity loss than fixing the same liability in a framework region.
Reported sequence liabilities
The block screens for the following predefined liabilities.
Sequence validity
| Liability | Motif | Fixability | Description |
|---|---|---|---|
| Stop codon | * | Disqualifying | Protein synthesis terminates prematurely. The sequence does not encode a viable antibody. |
| Out-of-frame | _ | Disqualifying | A frameshift mutation produces a completely incorrect amino acid sequence. Not an engineering problem — the sequence is invalid. |
Structural liabilities
| Liability | Fixability | Description |
|---|---|---|
| Missing cysteines | Structural | Conserved disulfide bonds are absent. These bonds hold the antibody's core structure together; missing ones cannot be restored by simple substitution. |
| Extra cysteines | Hard to fix | An unpaired cysteine will bond with other antibody molecules, causing aggregation. The rogue cysteine must be identified and removed, which often requires structural modeling and risks reshaping the CDR. |
Fixable liabilities
| Liability | Motif | Risk | Fixability | Description |
|---|---|---|---|---|
| Deamidation | N[GS] | High | Fixable | Asparagine converts to aspartate, introducing a charge change and structural distortion. The GS context accelerates the reaction. Fix: N→Q, but this position is often a CDR contact — expect affinity testing. |
| Fragmentation | DP | High | Fixable | The D–P peptide bond is susceptible to cleavage under acidic conditions. Fix: D→E or P→A, but proline often drives loop conformation — removing it can reshape the binding site. |
| Isomerization | D[DGHST] | High | Fixable | Aspartate isomerizes to isoaspartate, inserting an extra methylene group into the backbone and causing conformational change. Fix: D→E. |
| N-linked glycosylation | N[^P][ST] | High | Fixable | Unintended sugar attachment in the variable region causes binding heterogeneity and manufacturing inconsistency. Fix: mutate any of the three positions to break the motif. |
| Hydrolysis | NP | Medium | Fixable | The N–P bond is prone to hydrolysis. Fix: N→Q or P→A. |
| Fragmentation | TS | Medium | Fixable | β-elimination under alkaline conditions can cleave this bond. Fix: T→S/A or S→A. |
| Deamidation | N[AHNT] | Medium | Easily fixable | Slower deamidation context than N[GS]. Fix: N→Q, usually safe. |
| Tryptophan oxidation | W | Medium | Easily fixable | Exposure to light or reactive oxygen species oxidizes the tryptophan side chain, reducing binding. Fix: W→Y/F — conservative substitution, low affinity risk except at CDR3 C-terminus. |
| Methionine oxidation | M | Medium | Easily fixable | Oxidized methionine can reduce binding affinity and, in Fc positions, shorten serum half-life. Fix: M→L/I — standard conservative substitution. |
| Deamidation | [STK]N | Low | Easily fixable | Weak deamidation context driven by the preceding residue. Very low spontaneous rate. Fix: N→Q if desired. |
Integrin binding (off by default)
| Liability | Motif | Risk | Fixability | Description |
|---|---|---|---|---|
| Integrin binding | RGD|RYD|KGD|NGR|LDV|DGE|GPR | Low | Easily fixable | These motifs mimic natural integrin ligands and can cause off-target cell adhesion in vivo. Relevant primarily for therapeutic candidates; less critical at discovery stage. Disabled by default. |
Custom liabilities
You can define additional liability motifs beyond the predefined set. Custom liabilities are applied to the selected regions alongside predefined ones and participate in all four score columns.
Adding a custom liability in the UI
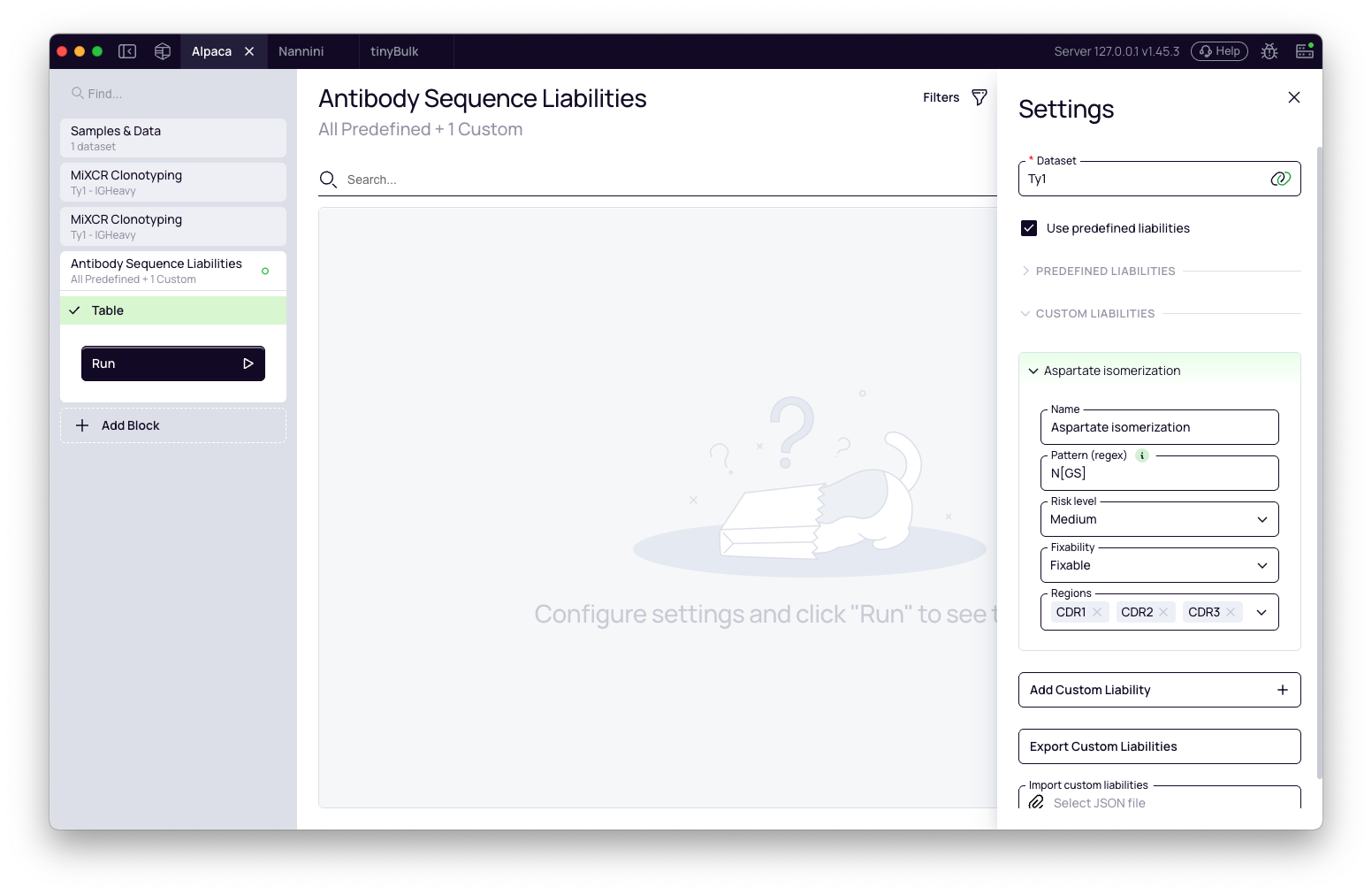
- In the block settings, click Add in the Custom liabilities section.
- Fill in the fields:
- Name — a unique label for this liability (must not match any predefined name)
- Pattern — a regex applied to the amino acid sequence (see Pattern syntax below)
- Risk level —
Low,Medium, orHigh - Fixability —
Easy fix,Fixable, orHard to fix - Regions — which regions to apply this motif to (default: CDR1, CDR2, CDR3)
Pattern syntax
Patterns are standard regular expressions applied to single-letter amino acid sequences.
| Construct | Example | Matches |
|---|---|---|
| Literal residue | W | Any tryptophan |
| Character class | [GS] | Glycine or serine |
| Negated class | [^P] | Any residue except proline |
| Sequence | DP | Aspartate followed by proline |
| Alternation | RGD|KGD | RGD or KGD |
Importing and exporting custom liabilities
For sharing or reusing a custom liability set across projects, use the Export and Import buttons in the custom liabilities section.
Export downloads your current custom entries as custom-liabilities.json. Import loads a JSON file and replaces the current custom entries, applying the same validation (duplicate names, invalid patterns, empty regions).
The JSON format is an array of objects:
[
{
"name": "My motif",
"pattern": "N[GS]",
"riskLevel": "High",
"fixability": "fixable",
"regions": ["CDR1", "CDR2", "CDR3"]
},
{
"name": "Integrin mimic (custom)",
"pattern": "RGD|KGD",
"riskLevel": "Low",
"fixability": "easily_fixable",
"regions": ["CDR1", "CDR2", "CDR3", "FR1"]
}
]
Field reference:
| Field | Type | Allowed values |
|---|---|---|
name | string | Any unique text; must not match a predefined liability name |
pattern | string | Valid regex in single-letter amino acid notation |
riskLevel | string | "Low" | "Medium" | "High" |
fixability | string | "easily_fixable" | "fixable" | "hard_to_fix" |
regions | array | One or more of "CDR1" "CDR2" "CDR3" "FR1" "FR2" "FR3" "FR4" |
To run only your custom set without the predefined liabilities, uncheck Use predefined liabilities in the settings panel.
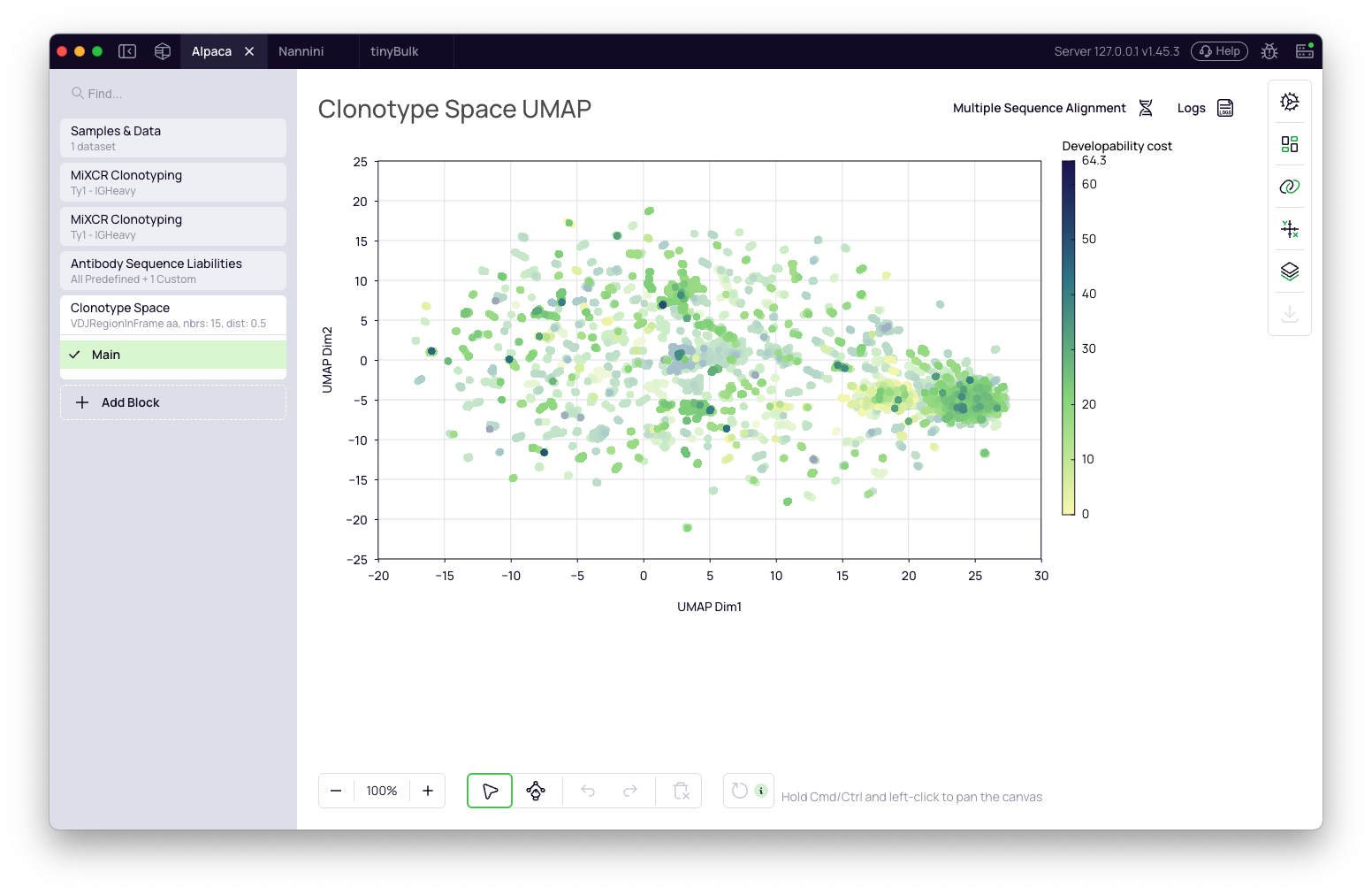
Downstream analysis: visualizing liabilities
The liability data can be used in downstream selection and visualization blocks. A common application is mapping liability scores onto the Clonotype Space UMAP to see how they relate to enrichment and sequence diversity.
- Go to your Clonotype Space block.
- Open the Data Mapping panel on the right.
- Use the Grouping / Color dropdown to select
Developability cost— this colors each point by its engineering effort score. - Use the Highlight dropdown to select
Developability riskand choose the values you want to emphasize, such asNone,Medium, andHigh, to surface candidates across the risk spectrum.
The screenshot below shows a UMAP colored by Developability cost with Developability risk highlighted for None, Medium, and High — making it easy to spot highly enriched clonotypes with low engineering burden.