How to create a custom reference library
Analyzing immune repertoire data from non-model organisms presents a unique challenge: the absence of built-in germline gene reference libraries. Accurate V(D)J gene alignment is crucial for clonotyping, but standard tools often only support common species like humans and mice.
Platforma solves this by allowing you to build a custom reference library directly within your workflow. This guide demonstrates how to create a custom reference library from your own FASTA files with gene sequences and use it to analyze data from any species.
You can also watch the full video tutorial here:
Step 1: Build a Custom Reference Library
This guide assumes you have already created a project and imported your raw sequencing data (e.g., FASTQ files) into a dataset. Our example uses IGH repertoire data from a dog.
The first step is to build the reference library that MiXCR clonotyping will use for alignment.
- Click Add Block to open the block library.
- Search for and select the MiXCR reference library builder block.
- Click Add To Project.
Now, configure the library builder in the Settings panel on the right.
- Species: Give your custom library a descriptive name. We'll use "dog".
- Chains: Select the immune receptor chains you are building the library for. In this example, we select IGH.
- Gene Segment Source: For each segment (V, J, and the optional D/C), you need to provide the gene sequences.
- Change the source from the default "From Built-in Species" to From Fasta File.
- Click Choose file and upload your FASTA file containing the sequences for that segment (e.g.,
v-genes.fa,j-genes.fa,d-genes.fa). - For the V segment, specify the Covered region. In most cases, this will be "V region," which assumes your sequences start at FR1.
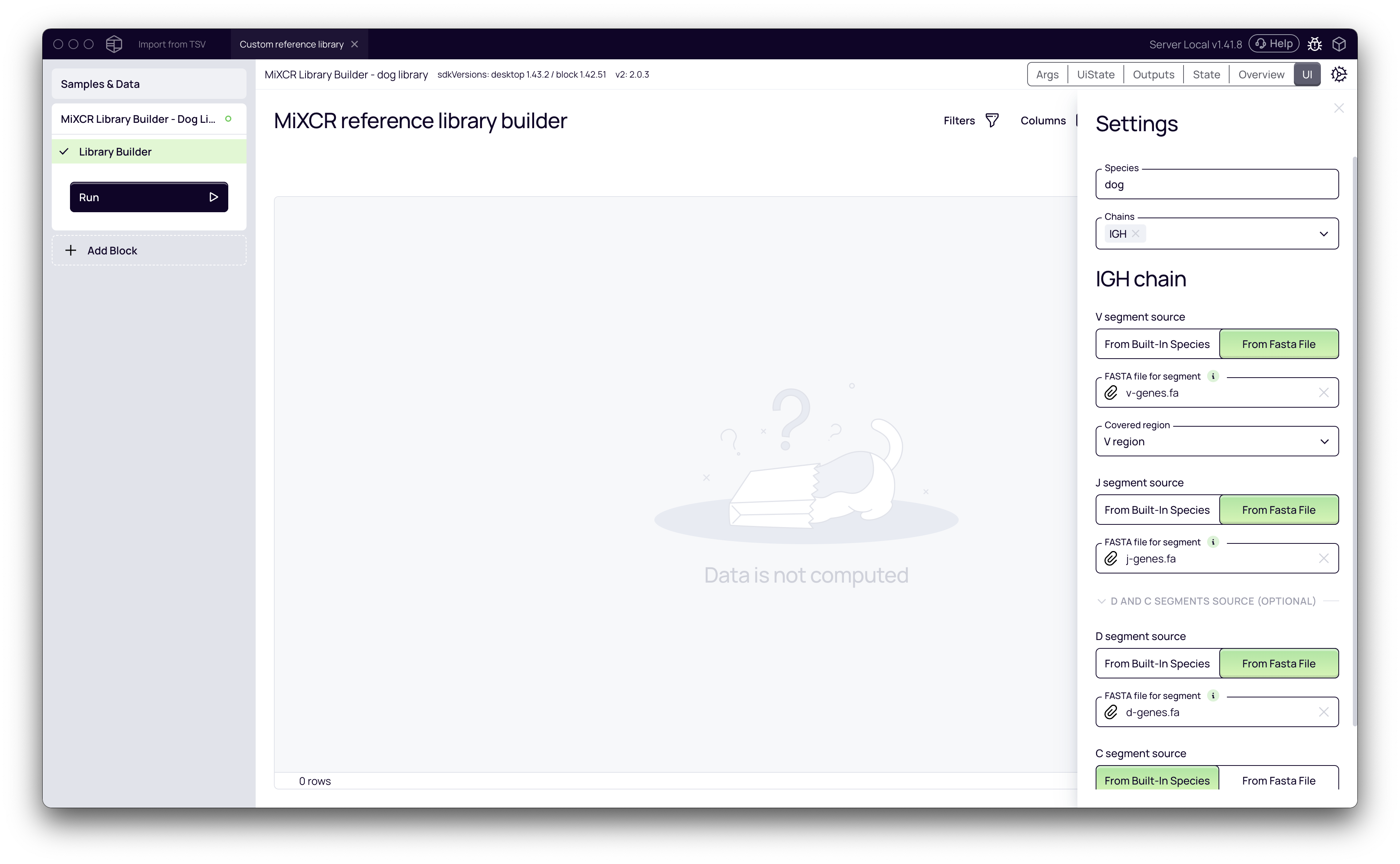
For the library to build correctly, your sequences must be fully defined. The FASTA files cannot contain any gaps or ambiguous nucleotide codes (like 'N').
- Once all settings are configured, click the Run button on the
Library Builderblock in the left panel. The block will process your FASTA files and construct a compatible reference library.
When the process is complete, you can view the resulting library table, which contains all the parsed gene segments.
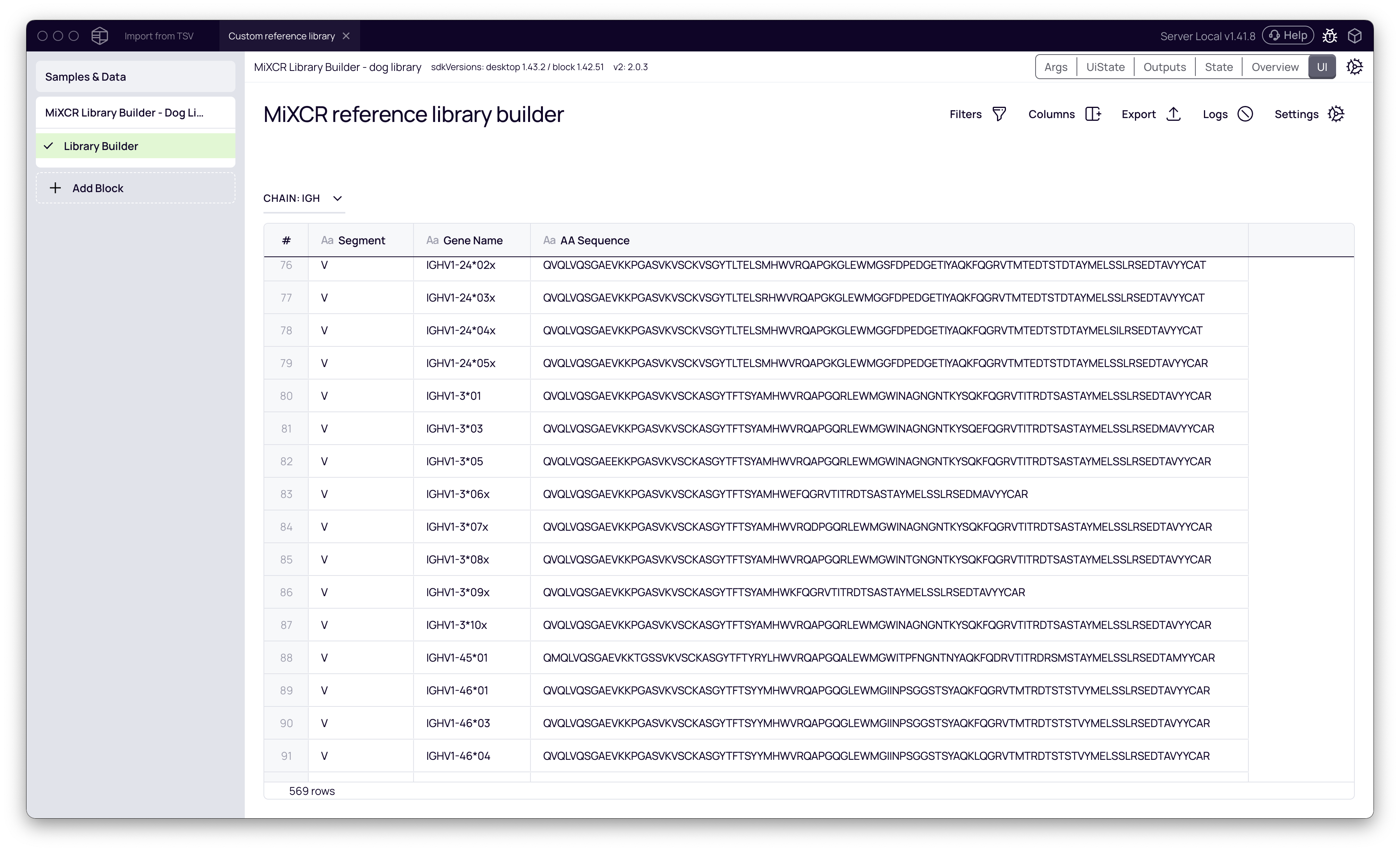
Step 2: Run Clonotyping with the Custom Library
With the custom reference library built, you can now perform clonotyping analysis.
- Click Add Block and add the MiXCR Clonotyping block to your project.
- Configure the clonotyping settings:
- Select dataset: Choose the dataset containing your sequencing data (e.g., "Custom dog").
- MiXCR Preset: Select a suitable preset based on your library preparation protocol.
- Receptors: Select the receptor type that matches your data and your custom library (e.g., IG Heavy).
- Expand the Advanced Settings section. This step is critical for using your custom library.
- Under Custom reference library, select From Library Builder.
- From the Custom library dropdown menu, choose the "dog" library you just created.
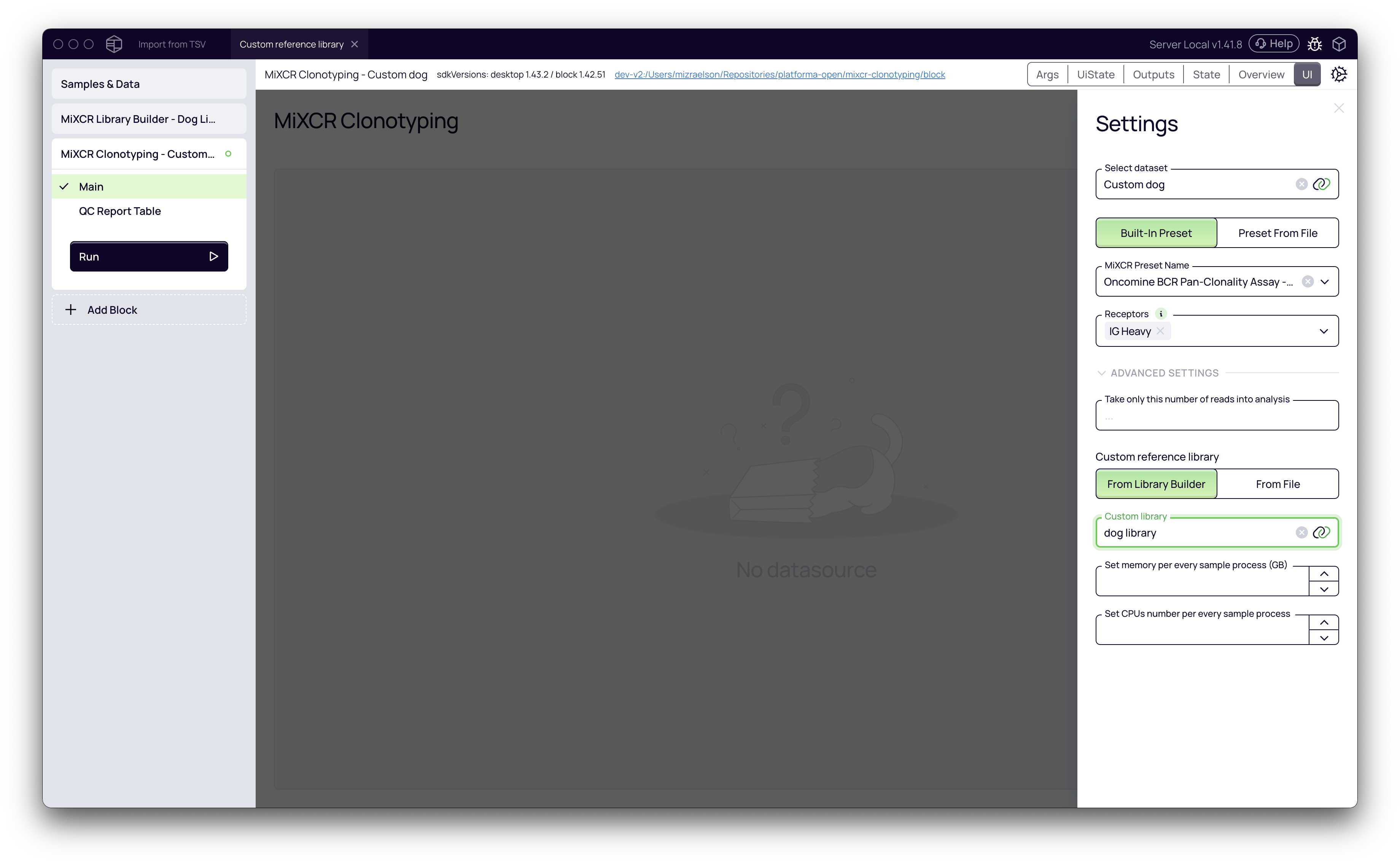
- Click Run to start the clonotyping analysis. MiXCR will now align the reads in your samples against the custom dog reference library.
Step 3: Review Results and Continue Analysis
Once the analysis is complete, you can inspect the results to confirm that the custom library worked correctly.
Click on the sample row in the MiXCR Clonotyping block to open the results. The Alignments chart shows the percentage of successfully aligned reads. A high alignment rate (e.g., >90%) indicates that the reference library was a good match for the data.
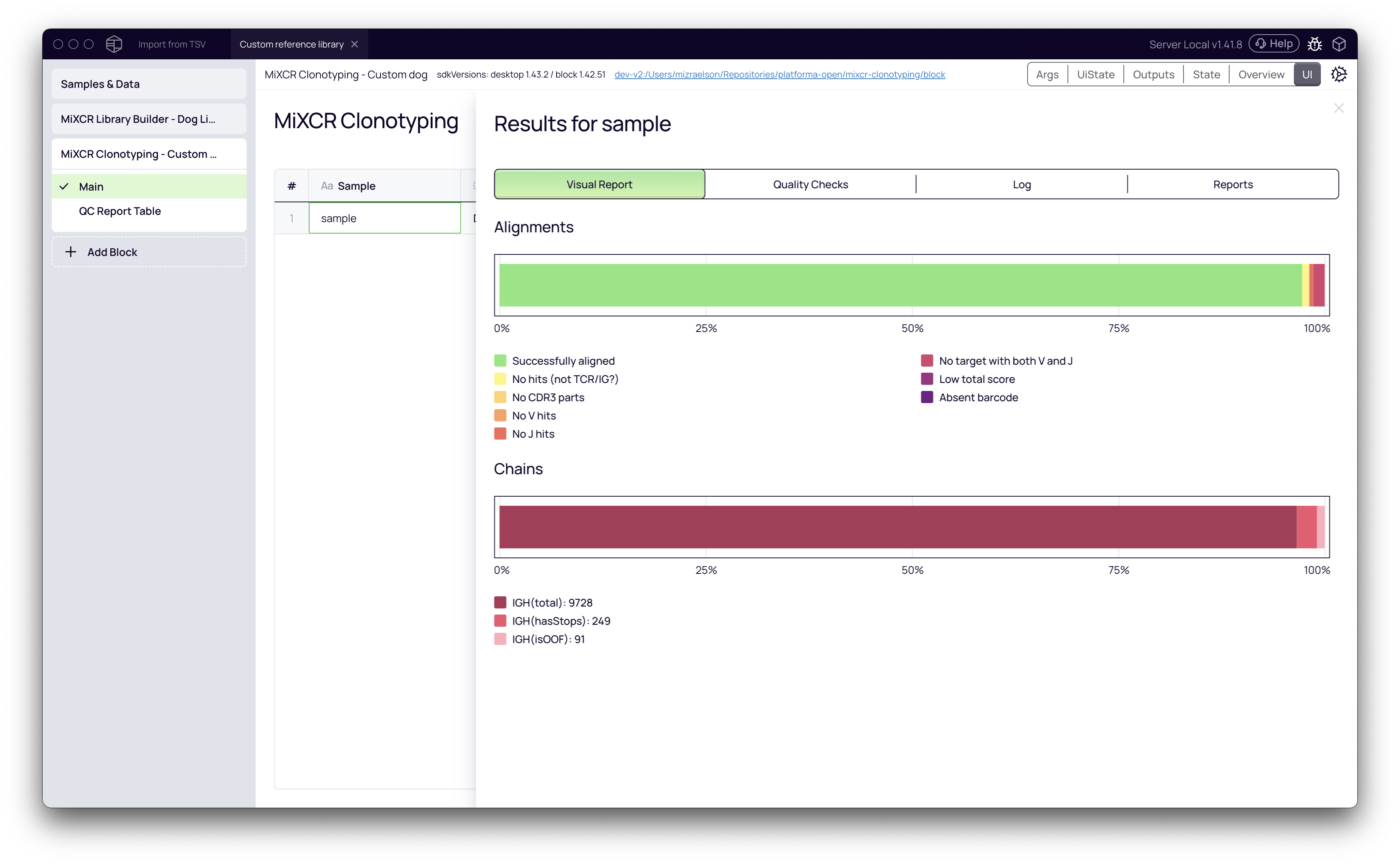
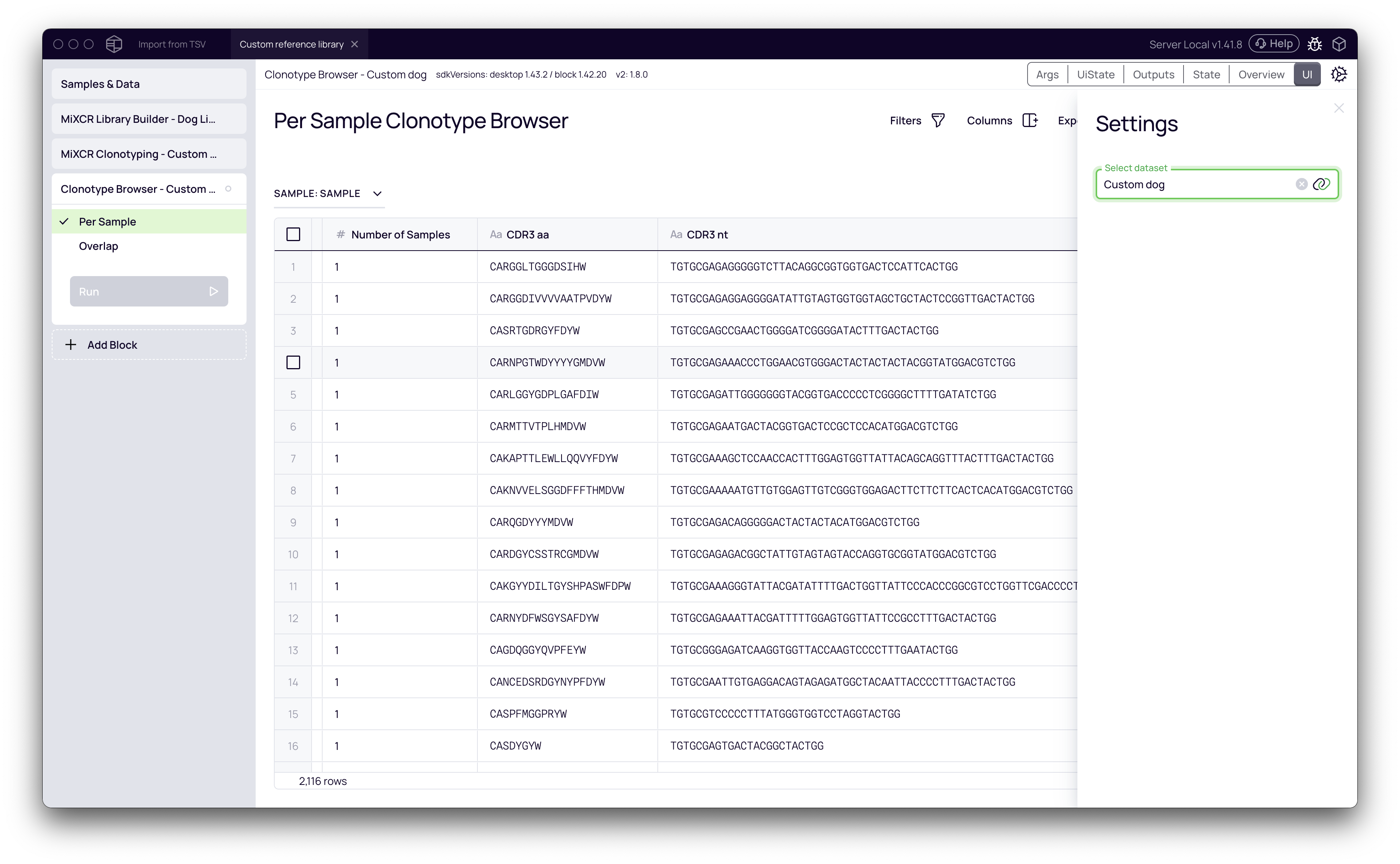
Your data, now annotated with custom V, D, and J gene information, is ready for any downstream analysis block in Platforma. For example, you can add a Clonotype Browser block to explore the resulting clonotypes.
Next Steps
Congratulations! You have successfully analyzed data from a non-model organism by building and applying a custom reference library. You can now proceed with deeper analyses, such as:
- Exploring clonotype diversity and overlap.
- Visualizing VJ gene usage.
- Performing differential abundance analysis based on your sample metadata.
This powerful feature opens the door to studying immune repertoires across a vast range of species, all within Platforma's code-free environment.